Hereditary Angioedema (HAE) is a rare, genetic disorder characterized by recurrent episodes of severe swelling (angioedema) in various parts of the body, including the skin, gastrointestinal tract, upper airway, and limbs.
Phase I
Systems Architecture
Status: In Progress
The Systems Architecture of Hereditary Angioedema is published as a web based tool open to public.
Phase II
Publications
Status: Open
A peer-reviewed paper from the Hereditary Angioedema will be published for the benefit of community.
Phase III
In Silico Modeling
Status: Open
In this phase, in silico modeling will develop mathematical models of Hereditary Angioedema pathogenesis.
Phase IV
Combination Screening
Status: Open
Combination Screening will be conducted to identify compounds targeting biological processes in Hereditary Angioedema pathogenesis.
Phase V
Patents
Status: Open
In this phase, a US patent will be filed for the newly discovered combination(s).
Phase VI
Licensing and Manufacturing
Status: Open
The Hereditary Angioedema Initiative will license and manufacture nutraceuticals to support treatment of Hereditary Angioedema.
Hereditary Angioedema (HAE) is a rare, genetic disorder characterized by recurrent episodes of severe swelling (angioedema) in various parts of the body, including the skin, gastrointestinal tract, upper airway, and limbs. Unlike typical allergic reactions, HAE is not caused by histamine release and does not respond to standard allergy treatments like antihistamines or corticosteroids.
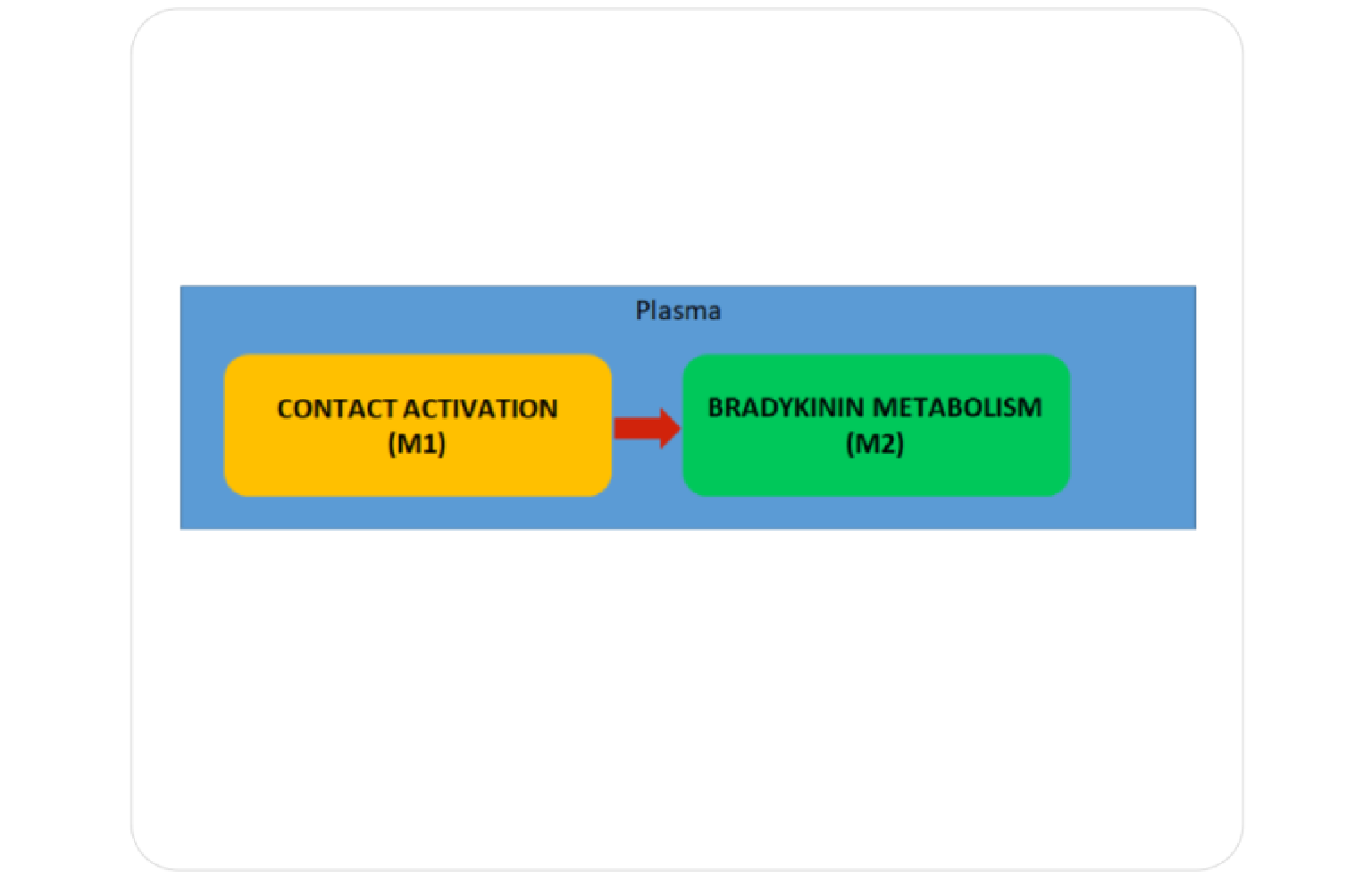
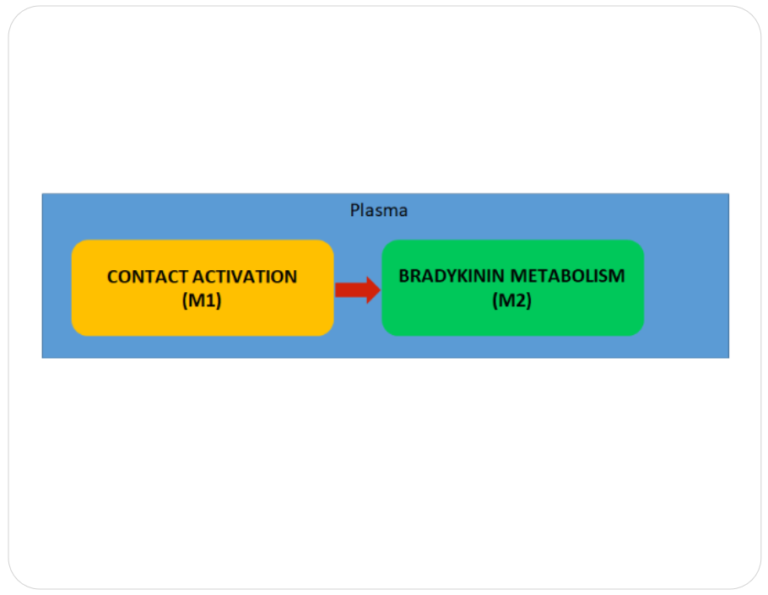
HAE is most commonly caused by a deficiency or dysfunction of C1 esterase inhibitor (C1-INH), a protein that helps regulate inflammation and the immune response. The deficiency leads to uncontrolled activation of the bradykinin pathway, which increases vascular permeability and results in fluid leaking into surrounding tissues, causing swelling. HAE is inherited in an autosomal dominant manner, meaning only one copy of the defective gene is sufficient to cause the disorder.
Symptoms often begin in childhood or adolescence and can worsen over time. Swelling attacks may last for several days and can be extremely painful, especially when affecting the abdomen. Swelling of the airway can be life-threatening if not treated promptly. Unlike other types of angioedema, HAE is not accompanied by hives.
Diagnosis involves blood tests to measure levels and function of C1-INH and complement proteins. Early and accurate diagnosis is critical to prevent complications and improve quality of life.
There is no cure for HAE, but several effective treatments are available. These include C1-INH replacement therapy, bradykinin receptor antagonists, and kallikrein inhibitors to prevent or treat acute attacks. With proper management, individuals with HAE can lead full, active lives.
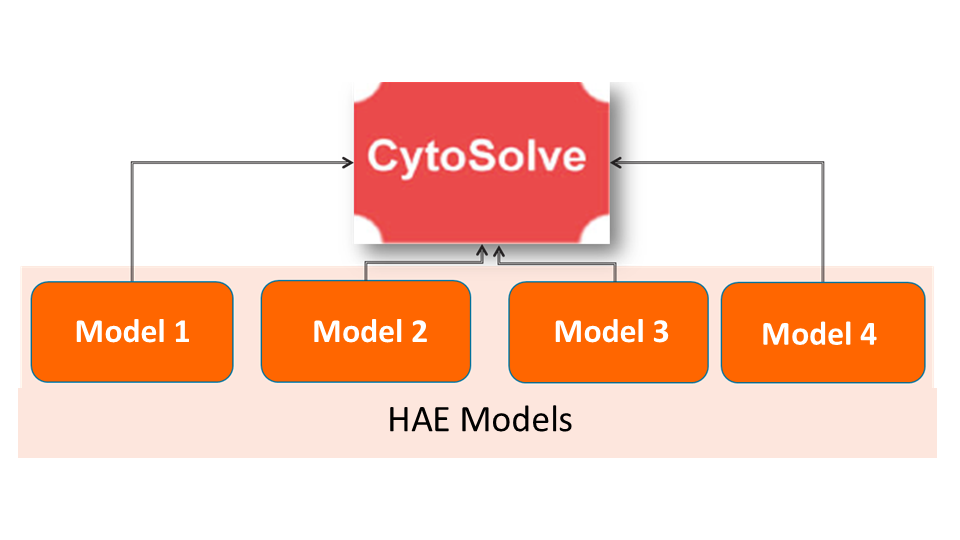
Systems Architecture
The Systems Architecture of Hereditary Angioedema is published as a web based tool open to public . Click below to interact with the Systems Architecture
A peer-reviewed publication resulting from the Hereditary Angioedema Research Initiative will be published in the reputed journal for the benefit of community. This phase is yet to begin.
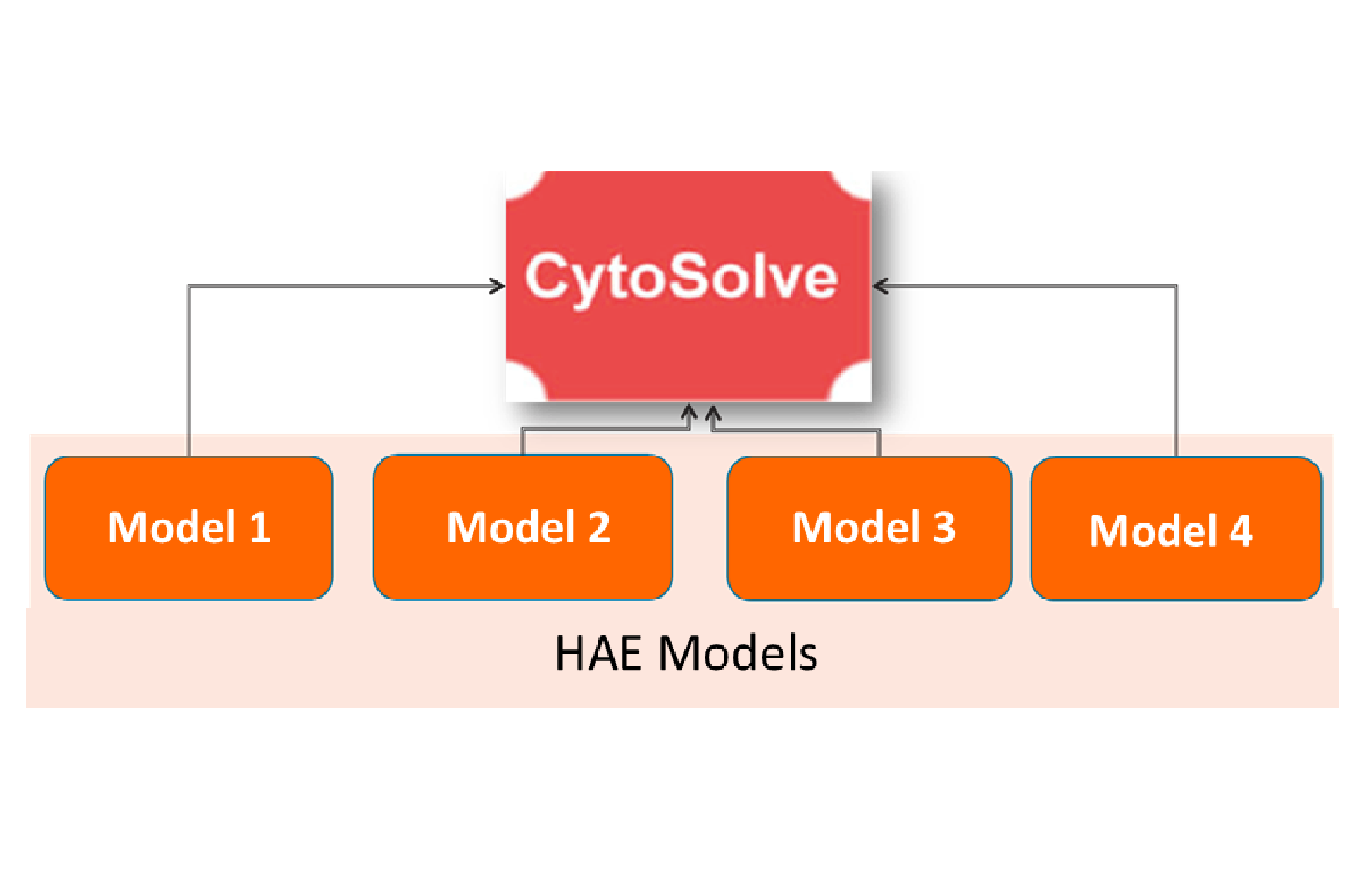
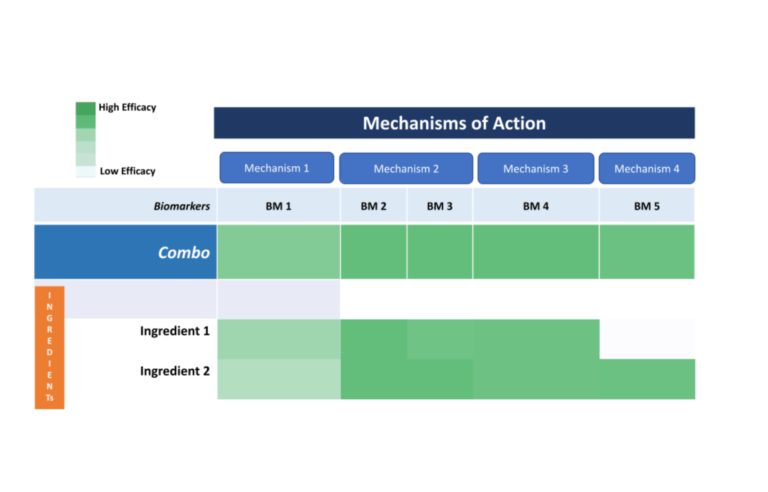
In this phase, the Hereditary Angioedema initiative will conduct in silico modeling to identify and test the efficacy of natural compounds against the Hereditary Angioedema diseases.
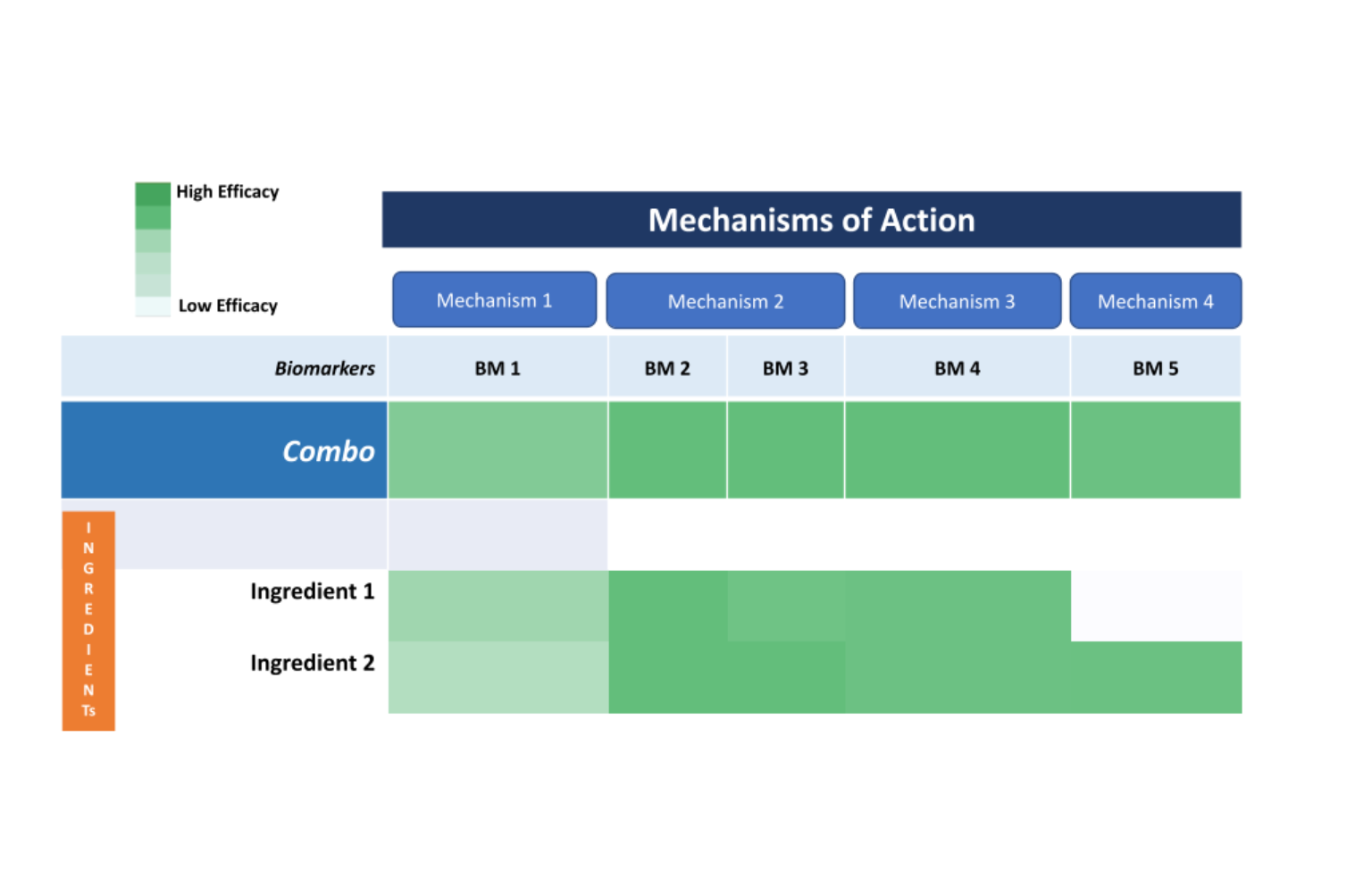
In this phase, combination screening will be performed to identify potential ingredient/compounds that target the biological process implicated in Hereditary Angioedema pathogenesis. This phase is yet to begin
The Open Science Institute® through its Hereditary Angioedema Initiative is moving towards getting patents for a revolutionary molecule that effectively combats HAE disease, contributing breakthrough real solutions to society worldwide.
The Hereditary Angioedema Initiative plans to discover, develop, license and manufacture proprietary nutraceuticals to support treatment of Hereditary Angioedema. Support our mission to bring this innovation to those who need it most. Please support this phase by donating to the Hereditary Angioedema Initiative